
中大新聞網(wǎng)訊(通訊員李民)中山大學(xué)藥學(xué)院李民課題組近日在權(quán)威期刊Redox Biology發(fā)表題為“Suppression of ATG4B by copper inhibits autophagy and involves in Mallory body formation”的研究論文,揭示了銅離子通過抑制ATG4B導(dǎo)致自噬失活,進(jìn)而阻礙威爾森病蛋白聚集體清除的一種新機制。
銅離子是人體必需的微量元素,但其過量蓄積會導(dǎo)致疾病發(fā)生,如威爾森病。威爾森病是ATP7B基因突變導(dǎo)致的常染色體隱性遺傳病,ATP7B基因突變使其編碼的ATP7B蛋白功能受損,肝細(xì)胞膽管側(cè)的ATP7B無法轉(zhuǎn)運銅離子至膽汁以排泄,過量蓄積的銅離子經(jīng)肝細(xì)胞血管測進(jìn)入血液循環(huán),引起全身多臟器損傷,其中肝臟和神經(jīng)系統(tǒng)病變最常見。其肝臟病理可表現(xiàn)為脂肪變性、炎癥浸潤、氣球樣變和Mallory小體生成等 。Mallory小體是細(xì)胞質(zhì)中的嗜酸性包涵體,在蛋白質(zhì)錯誤折疊和蛋白酶體功能受損等情況下可形成,主要成分為p62、泛素化蛋白和角蛋白K8/K18。前期研究表明激活自噬有助于Mallory小體的清除,但其具體機制尚不明確 。自噬是一種進(jìn)化保守的維持細(xì)胞穩(wěn)態(tài)的自我保護(hù)機制,已發(fā)現(xiàn)有四十余種ATG蛋白參與此過程 。其中半胱氨酸蛋白酶家族成員ATG4B可將未活化型的pro-LC3酶切為游離型的LC3-I,還可以將脂化型的LC3-II酶切回游離型LC3-I以循環(huán)利用。這兩步酶切對于自噬體的形成至關(guān)重要,因此抑制ATG4B會導(dǎo)致自噬功能失活。ATG4B是否通過調(diào)節(jié)自噬而影響銅離子蓄積導(dǎo)致形成Mallory小體或清除尚不清楚。
該研究首先發(fā)現(xiàn)銅離子在體外蛋白水平可有效且特異性地抑制ATG4B蛋白的酶活性,并在細(xì)胞水平靶向ATG4B而抑制自噬;銅離子可引起p62和ATG4B在細(xì)胞水平形成聚集體,且在細(xì)胞和體外蛋白水平誘導(dǎo)ATG4B發(fā)生寡聚化,該寡聚化現(xiàn)象受到氧化還原調(diào)控。這些蛋白聚集現(xiàn)象與威爾森病中銅離子引起Mallory小體的生化特征相似,并且在威爾森病的細(xì)胞模型中,過表達(dá)ATG4B可一定程度逆轉(zhuǎn)銅離子引起的自噬抑制以及相應(yīng)的Mallory小體形成。綜上,該研究發(fā)現(xiàn)了一種新的銅離子損傷機制,即銅離子靶向ATG4B而抑制自噬進(jìn)而促進(jìn)威爾森病中Mallory小體的生成并抑制其清除。該研究為全面認(rèn)識銅離子與自噬的關(guān)系以及在威爾森病發(fā)病機制中的作用提供了新的實驗證據(jù)。
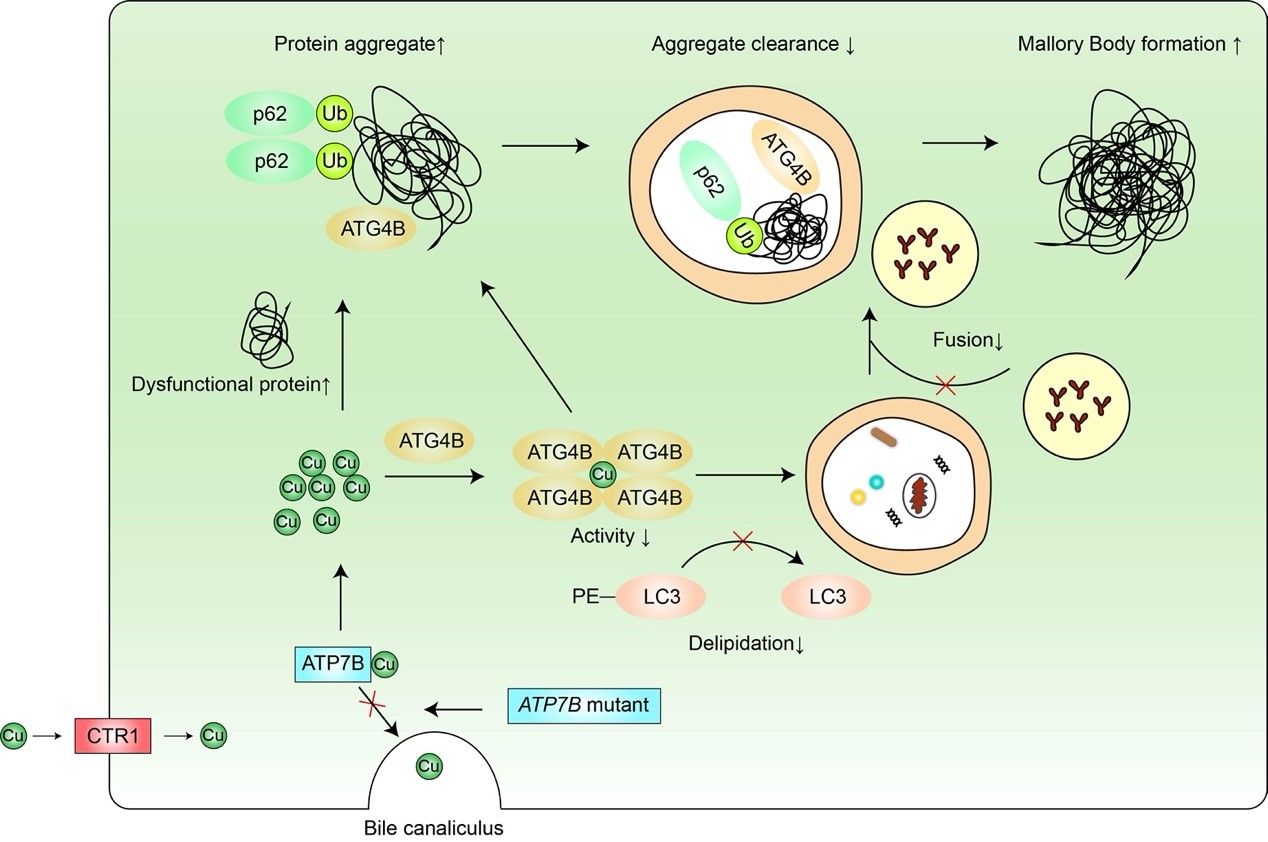
圖1. ATG4B參與銅離子蓄積形成Mallory小體形成示意圖
這項工作也是李民課題組前期發(fā)現(xiàn)在過氧化氫誘導(dǎo)條件下ATG4B氧化還原調(diào)控位點Cys292和Cys361研究工作的基礎(chǔ)上( The protease activity of human ATG4B is regulated by reversible oxidative modification, Autophagy, 2020, 16: 1838-1850),新發(fā)現(xiàn)的一種銅離子介導(dǎo)的氧化還原調(diào)控模式。
中山大學(xué)藥學(xué)院碩士生夏凡、博士后伏園園和碩士生謝華中為論文的共同第一作者,李民為通訊作者。中山大學(xué)藥學(xué)院劉培慶團(tuán)隊和動物中心張薇團(tuán)隊為這項工作提供大力支持。該研究得到國家自然科學(xué)基金、廣東省自然科學(xué)基金以及廣東省手性分子與藥物發(fā)現(xiàn)重點實驗室的資助。



