
近日,我校附屬第六醫院吳小劍主任醫師、王文宇副研究員聯合新加坡基因組研究院于強教授團隊在Nature Communications雜志在線發表了題為“Stromal induction of BRD4 phosphorylation Results in Chromatin Remodeling and BET inhibitor Resistance in Colorectal Cancer”的研究論文。該研究成果揭示了結直腸腫瘤微環境中的炎性因子IL-6/IL-8通過激活下游JAK2激酶,促進BRD4酪氨酸97/98位磷酸化,增強結直腸腫瘤對BET抑制劑的抵抗以及腫瘤惡性程度的表觀遺傳調控新機制,為聯合靶向腫瘤微環境IL6/JAK2信號及BRD4治療CRC的新策略提供了強有力的理論依據。
附屬第六醫院王文宇副研究員和新加坡基因組研究院Yen-An Tang博士為論文的共同第一作者;新加坡基因組研究院于強教授,附屬第六醫院吳小劍主任醫師和王文宇副研究員為本文共同通訊作者。該研究得到國家自然科學基金、中山大學青年教師重點培育計劃、廣東省重點研發計劃、廣東省科技計劃等項目的資助。
BET 家族蛋白,特別是 BRD4,作為重要的轉錄和表觀遺傳調節因子,與包含結直腸腫瘤在內的多種腫瘤進展密切相關,已成為科學家們開發癌癥創新療法的新興靶點之一。目前已有多個小分子BET抑制劑(BETi,BET inhibitors)進入臨床試驗,雖然在血液腫瘤早期臨床實驗中BETi顯示出令人鼓舞的效果,但在實體腫瘤中療效卻差強人意。
近年來,“腫瘤生態學說”逐漸成為科學家們試圖攻克癌癥的突破口,腫瘤微環境和腫瘤細胞被認為是滋生腫瘤的“土壤”和“種子”,腫瘤細胞和微環境中其他組分的頻繁互動被認為是導致腫瘤進展、耐藥的關鍵因素。
炎癥微環境是腫瘤的十大特征之一,既往研究表明炎性因子可以通過激活關鍵促腫瘤信號通路或抑制抗腫瘤免疫應答而促進腫瘤發生發展,但關于炎性因子是否能夠通過表觀遺傳調控(如染色體重塑)的方式參與調控腫瘤的相關生物學行為目前尚缺乏報道。
團隊人員首先建立了患者來源的腫瘤細胞(patient-derived cancer cells, PDCs)及腫瘤相關成纖維細胞(cancer-associated fibroblasts,CAFs)共培養模型。進一步通過細胞因子、表觀遺傳因子篩選聯合質譜分析等手段,鑒定出CAFs分泌的細胞因子IL-6/IL-8通過激活JAK2激酶通路,磷酸化BRD4,促進其與去泛素化酶UCHL3結合從而增強BRD4蛋白穩定性。此外,第97位酪氨酸(Y 97)是BRD4 與BET 抑制劑-JQ1 結合口袋中的關鍵殘基,BRD4 Y97/98 的磷酸化削弱了 JQ1與BRD4的結合能力,導致 JQ1治療抗性。更深入的探索發現 BRD4磷酸化還有助于其與 STAT3 的結合,誘導染色體重塑,協同促進原癌基因增強子活性及相關基因的轉錄。抑制 IL-6/IL-8-JAK2 信號能夠消除 BRD4 磷酸化,增加BETi敏感性。
更重要的是,該團隊利用來自附屬第六醫院臨床結直腸腫瘤患者的組織芯片證實了CAFs-JAK2信號通路激活與BRD4磷酸化水平呈現顯著正相關,并與病人的預后、腫瘤轉移相關。同時,進一步研究利用腫瘤異種移植模型評估了IL-6/IL-8-JAK2-BRD4通路作為結直腸腫瘤治療靶點的效果。實驗結果顯示,與單藥相比,IL-6/IL-8通路抗體/抑制劑(Tocilizumab/Reparixin)或JAK2抑制劑Pacritinib與BETi聯用對CAFs誘導的BETi抗性具有很好的抑制作用,顯著抑制結直腸腫瘤生長。
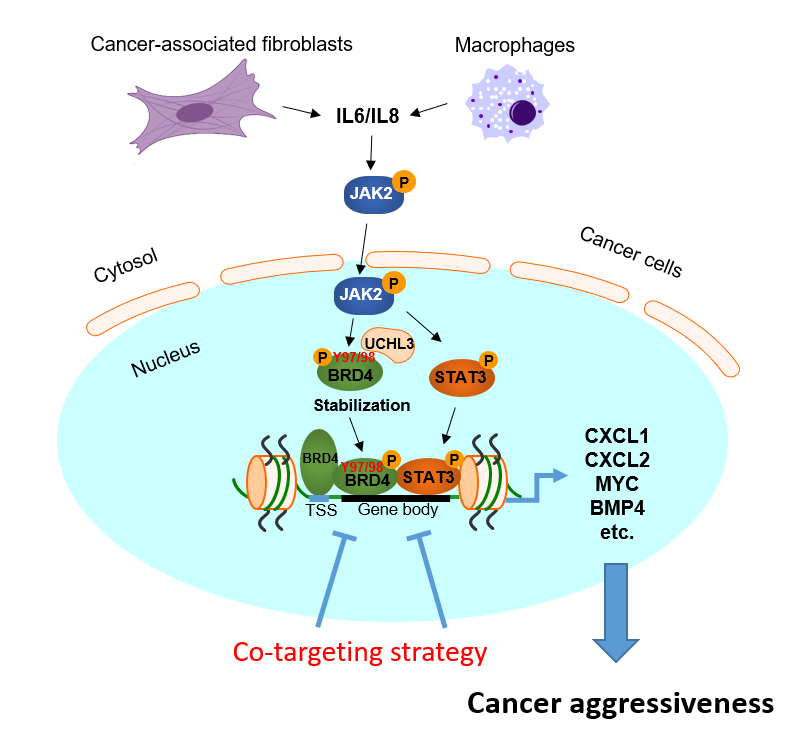
IL-6/8-JAK2-BRD4信號軸調控BET抑制劑耐藥的模式示意圖
該研究展示了腫瘤微環境通過“三管齊下”的方式導致BETi耐藥的新機制,即增強BRD4蛋白穩定性;削弱其與BET抑制劑結合能力;誘導染色體重塑,促進原癌基因增強子活性,最終導致結直腸腫瘤對BETi的抵抗及腫瘤惡性程度增加。該發現強調了腫瘤微環境中的炎癥信號通過染色體重塑的方式實現與腫瘤細胞的“對話”,為理解結直腸腫瘤的進展、耐藥提供了新見解。研究提出的聯合靶向IL-6/IL-8/JAK2 及其活化的 BRD4可成為臨床轉化的選擇。評估 pJAK2、pBRD4 和 UCHL3 在 CRC 中的表達可能有助于指導患者選擇合適的治療策略,為結直腸腫瘤患者的治療提供了新方向。


