
2021年8月11日,Journal of Experimental & Clinical Cancer Research雜志在線發表我校附屬第三醫院吳斌教授團隊題為“Phosphorylation of NF-κBp65 drives inflammation-mediated hepatocellular carcinogenesis and is a novel therapeutic target”的長篇研究論文(中科院一區)。
該研究發現肝臟慢性炎癥反應,尤其是TNF-α,誘發肝細胞NF-κBp65表達上調,進一步通過β-arrestin1介導NF-κBp65-Ser536位點磷酸化,繼而激活下游Akt/mTOR信號通路,促進了肝癌的發生與發展。該研究是吳斌教授團隊在Nature Communications發表題為“β-arrestin1 enhances hepatocellular carcinogenesis through inflammation-mediated Akt signalling”及在Autophagy發表題為“HBx induces hepatocellular carcinogenesis through ARRB1-mediated autophagy to drive the G1/S cycle”研究工作的延續及深入。
該團隊在人肝癌組織樣本及血液樣本中檢測發現NF-κB信號通路5個亞基NF-κB1、NF-κB2、p65(RelA)、RelB、c-Rel中尤以p65(RelA)的表達及磷酸化最顯著。根據這個重要臨床發現,該團隊進一步通過肝細胞特異敲除(L- NF-κBp65 KO)小鼠構建肝癌模型,與野生型小鼠比較,L-NF-κBp65基因敲除后肝癌發生的數量、體積等均顯著低于野生型小鼠。在DEN、四氯化碳、TNF-α構建的3種小鼠肝臟炎癥模型中,NF-κBp65的表達水平及磷酸化均顯著升高;肝癌細胞實驗及裸鼠成瘤實驗中,NF-κB信號通路抑制劑(Bay 11-7082、PDTC)明顯抑制肝癌細胞的增殖及移植瘤的生長。進一步通過β-arrestin1基因敲除小鼠、肝癌細胞β-arrestin1基因沉默實驗、及NF-κBp65磷酸化位點突變實驗等進行深入的機制研究,發現β-arrestin1顯著激活NF-κBp65-Ser536位點的磷酸化,但并不激活Ser276或Ser529位點的磷酸化,而Ser536位點的磷酸化活化了下游Akt/mTOR信號通路,結果誘發肝細胞的癌變及促進肝癌的發展(圖1)。
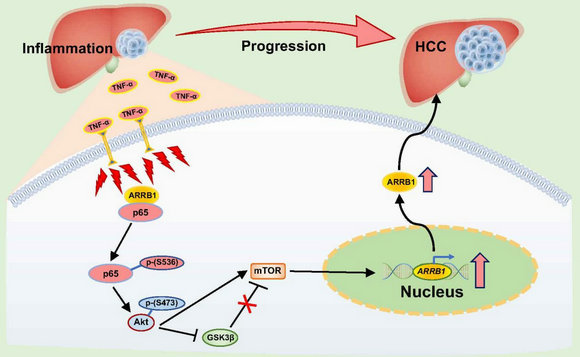
圖1:在肝臟炎癥微環境中,TNF-α激活NF-κB信號通路,在β-arrestin1(ARRB1)誘導下NF-κBp65亞基Ser536位點磷酸化上調,從而活化下游Akt/mTOR信號通路,以誘發肝細胞的癌變及促進肝癌的發展
此項研究為理解肝臟炎癥環境中NF-κBp65的顯著上調及磷酸化在肝癌中的作用及相關機制提供了新的科學依據,同時揭示了β-arrestin1誘導NF-κBp65-Ser536磷酸化的新機制,為NF-κBp65及其Ser536位點磷酸化作為肝癌防治的關鍵藥物靶點提供了新的理論基礎。
吳斌教授的學生徐璇博士與雷一鳴博士為此文的共同第一作者,吳斌教授和楊逸冬副主任醫師為共同通訊作者,我校附屬第三醫院為論文第一作者單位及通訊作者單位。該研究獲得國家自然科學重點項目及面上項目(U1501224與82070574)、廣東省自然科學基金團隊項目(2018B030312009)的支持。
論文鏈接:https://jeccr.biomedcentral.com/articles/10.1186/s13046-021-02062-x



