
中大新聞網訊(通訊員張銳)近年來,在轉錄組中發現的各種RNA編輯修飾促進了表觀轉錄組學領域的迅速發展。這些RNA編輯修飾位點在調控RNA代謝的各個層面發揮著關鍵作用,并廣泛涉及到多種生物過程,具備重要的功能。例如,m5C修飾在胚胎發育、腫瘤發展和病毒調控中扮演著重要角色,而m6A修飾和A-to-I編輯酶在多種癌癥中異常調節,被認為是有潛力的癌癥治療靶點。
與此同時,單堿基分辨率的測序技術在飛速發展。針對不同的RNA編輯修飾,研究人員發明了若干基于化學方法的檢測手段,比如:Bisulfite sequencing (5-甲基胞嘧啶修飾, m5C) ;CMC-seq, BID-seq(假尿嘧啶修飾,ψ);GLORI,eTAM-seq (6-甲基腺嘌呤修飾, m6A)。除了化學方法,使用納米孔(Nanopore)直接進行RNA 編輯修飾測序的技術也方興未艾。但是,這些檢測手段往往伴隨著假陽性信號,而往往沒有很好的先驗知識去評估信號的真實性。比如,在Bisulfite sequencing中,由于RNA二級結構會妨礙脫氨基反應的進行,Bisulfite sequencing中往往存在大量的位于高GC含量區域的假陽性信號;這些假陽性信號和真實的具有特定基序(motif)的m5C位點混合在一起而難以分辨。同時,針對不同的測序方法,除了使用統計學參數外,也很難直接對它們進行比較。因而,當前需要一個技術手段進行RNA編輯修飾序列特征的比較和分類。
傳統的序列分析工具通常基于序列出現的頻率進行統計,從而獲得高頻出現的序列特征(即motif)。這些分析工具(如MEME,HOMER)為發現轉錄因子結合位點(TFBS)而設計——對于RNA修飾的motif發現并沒有進行優化。與TFBS的motif發現的情境不同,RNA修飾位點的序列是高度對齊的,且motif的長度往往很短。實際上,RNA編輯修飾motif的發現與單細胞測序中的可視化流程十分相似:RNA編輯修飾motif可以通過One Hot encoding轉化成高維向量,而這些高維向量可以通過Manifold法進行分解,(如UMAP,t-SNE)投影在二維平面上——與motif這一概念一致,如存在反復出現的相似的序列,它們將在二維投影的某個區域形成富集(高密度區域)。若能使用某種手段對投影進行聚類,并提取這些富集區域,就能夠以可視化的形式對給定序列進行分類以及motif的發掘。
基于以上原理,中山大學生命科學學院張銳課題組開發了一個基于非線性降維技術和密度聚類,稱為交互式RNA修飾motif可視化和亞型分類(iMVP,interactive epitranscriptomic Motif Visualization and Subtype Partition)的計算框架。該開源框架iMVP(https://github.com/SYSU-zhanglab/iMVP)能夠用于RNA修飾motif的去噪、亞型分類和可視化。與傳統方法相比,它在各種高通量數據、人工模擬高噪聲數據、超大數據集上都有出色表現。

圖1. iMVP框架
研究團隊運用iMVP工具對不同物種和發育時期的mRNA m5C圖譜進行了全面分析。他們不僅確認了已知的m5C motif,更意外地發現了兩種與酵母25S rRNA C2278和C2870 m5C位點相似的motif。這兩個位點在酵母中分別由Rcm1(NSUN5)和Nop2酶甲基化,因此作者合理地推測這兩種酶可能是m5C修飾的新writer。通過在HeLa 細胞中進行敲低實驗,作者成功驗證了這一假設,確定了NSUN5與Nop2是mRNA m5C修飾的兩個新writer。這一新發現,凸顯了 iMVP作為一種有效的工具,用于發現新的RNA修飾模式和識別新的修飾酶。這將有助于更深入地理解RNA修飾的復雜性和功能。
目前已經開發了多種生化方法,可以在單堿基分辨率繪制m6A/m6Am修飾在轉錄組中的分布。然而,不同方法之間的位點識別存在差異,因此需要對這些方法進行系統評估和比較。iMVP的出現填補了這一知識空白。研究團隊匯總了來自CIMS,CITS,m6ACE-seq, m6A-label-seq, MAZTER-seq, m6A-REF-seq,xPore和DART-seq,總共8種不同m6A/m6Am測序方法的數據,發現盡管這些方法都使用相同的細胞類型,但只有少數m6A和m6Am位點在不同方法之間重疊。這表明每種方法可能只捕獲了甲基化位點的部分子集。除外,該研究還評估了非抗體方法在m6A/m6Am測序中的可靠性。結果表明,m6A-label-seq和MAZTER-seq是目前最可靠的方法,為研究人員選擇合適的非抗體方法提供了重要的參考。
Nanopore測序數據存在修飾信號相位錯配的問題,限制了其在RNA編輯修飾位點的準確識別。為應對這一挑戰,研究團隊提出了相位匹配策略,成功解決了這一問題,使iMVP工具能夠更精確地識別RNA修飾位點。此外,研究人員還分析了ModTect數據集,其中包含了從RNA-seq數據中推測的大量RNA修飾位點。鑒于這些位點的復雜性和噪聲,需要一種可靠的篩選方法來鑒定真正的RNA修飾候選位點。為此,作者引入了"spiked iMVP"策略,通過將已知修飾信號的k-mers加入變異數據中,標記已知RNA修飾的模體偏好,并成功識別了高置信度的m1A、m1acp3Ψ和m22G位點。這些策略成功的擴展了iMVP工具的應用范圍
傳統的motifs 搜索工具通常僅適用于小規模數據集,而iMVP通過引入UMAP和HDBSCAN技術,并且通過使用GPU加速,成功應對了處理極大RNA修飾位點數據集的挑戰。研究團隊使用iMVP工具進行了對包含1500萬個A-to-I RNA編輯位點進行分析。他們觀察到不同類型的A-to- I RNA編輯位點在Alu、非Alu重復和非重復區域中呈現出截然不同的模式。進一步應用iMVP工具,他們獲得了高分辨率的數據結果,成功識別出各類編輯位點的簇群。
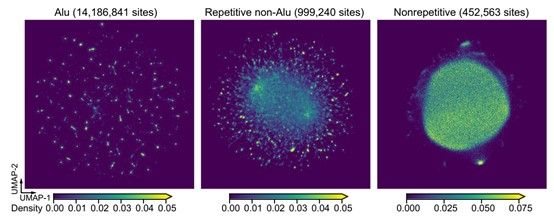
圖2. A-to- I RNA編輯位點在Alu、非Alu重復和非重復區域中呈現出截然不同的模體模式
總之,iMVP的開發為RNA編輯修飾研究帶來了新的可能性,為科研人員提供了一個更全面、更有效的工具,有望有助于更深入地理解RNA編輯修飾的復雜性和功能。
該成果于近期以“Epitranscriptomic subtyping, visualization, and denoising by global motif visualization”為題發表在Nature Communications。中山大學生命科學學院張銳教授,博士生劉健恒(現為康奈爾大學博士后)為本文的共同通訊作者,劉健恒、黃濤、姚靜為本文的并列第一作者,趙天璇、張鈺森也對本工作做出重要貢獻。中山大學生命科學學院為第一作者單位。


