
中大新聞網(wǎng)訊(通訊員崔雋)蛋白質(zhì)是組成生命的物質(zhì)基礎(chǔ),是生命活動(dòng)的主要承擔(dān)者,然而蛋白質(zhì)在合成之后,往往需要經(jīng)過一系列蛋白質(zhì)翻譯后修飾(Protein post-translational modifications, PTMs),通過功能基團(tuán)的添加或去除,調(diào)節(jié)蛋白質(zhì)的生化性質(zhì)從而改變其活性。近日,中山大學(xué)生命科學(xué)學(xué)院崔雋課題組在Molecular Cell雜志83期上連續(xù)發(fā)表兩篇學(xué)術(shù)論文,分別揭示了蛋白棕櫚酰化和焦磷酸化這兩種新型PTM對(duì)固有免疫應(yīng)答的關(guān)鍵調(diào)控作用機(jī)制。
蛋白棕櫚酰化介導(dǎo)自噬和NLRP3炎癥小體互作調(diào)控炎癥及其相關(guān)疾病的新機(jī)制
NLRP3炎癥小體是固有免疫系統(tǒng)的重要組成部分,其功能紊亂與自身炎癥性疾病、神經(jīng)性退行性疾病以及癌癥的發(fā)生發(fā)展密切相關(guān)。NLRP3突變引起炎癥小體的過度激活是早發(fā)型炎癥性腸病(VEOBD)、幼年特發(fā)性關(guān)節(jié)炎(JIA)和冷吡啉相關(guān)周期性綜合征(CAPS)等自身炎癥疾病的重要危險(xiǎn)因素,因此其活化必須被精細(xì)調(diào)控以維持免疫穩(wěn)態(tài)。崔雋課題組發(fā)現(xiàn)S-棕櫚酰化通過介導(dǎo)分子伴侶介導(dǎo)的自噬,在調(diào)控NLRP3蛋白穩(wěn)定性中發(fā)揮重要功能,并揭示S-棕櫚酰化缺陷的NLRP3突變與自身炎癥性疾病的發(fā)生具有一定的相關(guān)性。
S-棕櫚酰化是一種新型的蛋白翻譯后修飾,參與許多免疫相關(guān)蛋白質(zhì)的轉(zhuǎn)運(yùn)、定位和穩(wěn)定性的調(diào)節(jié)。然而,S-棕櫚酰化是否在調(diào)控NLRP3炎癥小體激活過程中發(fā)揮關(guān)鍵作用尚有待闡明。該研究結(jié)合S-棕櫚酰化抑制劑2-bromopalmitate(2BP)處理、酰基-生物素交換、S-棕櫚酰化修飾定點(diǎn)突變、蛋白互作質(zhì)譜、過表達(dá)/敲除棕櫚酰轉(zhuǎn)移酶ZDHHC12等實(shí)驗(yàn),證實(shí)ZDHHC12可以明顯促進(jìn)NLRP3的棕櫚酰化修飾,而其酶活缺失突變ZDHHC12 C127S則不能,表明ZDHHC12促進(jìn)NLRP3的棕櫚酰化修飾依賴于其酶活性。進(jìn)一步的研究發(fā)現(xiàn)Zdhhc12缺陷可在小鼠體內(nèi)促進(jìn)NLRP3炎癥小體的激活和IL-1β的釋放,從而加重Alum誘導(dǎo)的腹膜炎以及LPS誘導(dǎo)的膿毒血癥,揭示ZDHHC12在機(jī)體中發(fā)揮防止NLRP3炎癥小體過度激活的重要作用。通過抑制劑實(shí)驗(yàn),研究人員發(fā)現(xiàn)溶酶體抑制劑NH4Cl和CQ能夠顯著抑制ZDHHC12對(duì)NLRP3的降解作用。進(jìn)一步研究發(fā)現(xiàn)ZDHHC12介導(dǎo)的S-棕櫚酰化修飾通過增強(qiáng)NLRP3與分子伴侶HSC70的結(jié)合,促進(jìn)NLRP3進(jìn)入溶酶體進(jìn)行降解,最終抑制NLRP3炎癥小體的激活。
為了探究自身炎癥性疾病相關(guān)的NLRP3突變是否與棕櫚酰化失調(diào)相關(guān),研究人員篩選了一系列NLRP3突變并發(fā)現(xiàn) NLRP3的D21H、S102L、R490K、和G571R突變可以通過抑制NLRP3的S-棕櫚酰化修飾以增強(qiáng)其穩(wěn)定性,從而促進(jìn)炎癥反應(yīng)。進(jìn)一步的機(jī)制研究表明,這是由于NLRP3的相應(yīng)突變降低了NLRP3與ZDHHC12的結(jié)合,從而抑制HSC70對(duì)NLRP3的識(shí)別和降解。由于NLRP3 的S102L突變?cè)趤喼奕巳褐蓄l率較高,且在JIA和CAPS等自身炎癥性疾病患者體內(nèi)被多次發(fā)現(xiàn),該研究將有助于 NLRP3突變相關(guān)的自身炎癥性疾病患者的診斷與精確治療。鑒于S-棕櫚酰化修飾對(duì)NLRP3蛋白激活和穩(wěn)定性的重要調(diào)控作用,特異靶向棕櫚酰轉(zhuǎn)移酶的藥物可能是治療自身炎癥性疾病的理想靶標(biāo)。
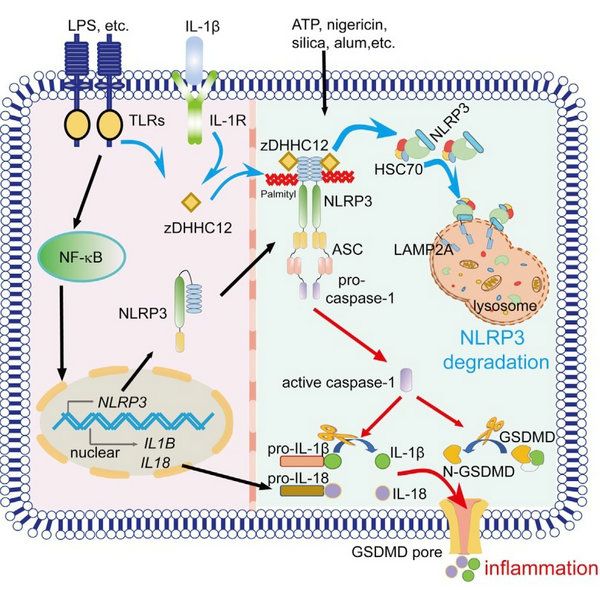
圖1 ZDHHC12通過棕櫚酰化,誘導(dǎo)NLRP3通過分子伴侶介導(dǎo)的自噬降解,抑制過度炎癥反應(yīng)的工作模型
這篇研究論文的第一作者是中山大學(xué)生命科學(xué)學(xué)院王麗邱博士,她的主要研究方向?yàn)檠装Y小體的免疫調(diào)控機(jī)制,于2022年入選博士后創(chuàng)新人才支持計(jì)劃,并在Nature Communications、Cellular & Molecular Immunology等雜志發(fā)表多篇一作文章。崔雋教授為該論文的通訊作者。該研究得到國(guó)家自然科學(xué)基金和博士后創(chuàng)新人才支持計(jì)劃等經(jīng)費(fèi)支持。
蛋白焦磷酸化調(diào)控抗病毒固有免疫的新機(jī)制
磷酸化修飾是目前已知分布最為廣泛的PTM。蛋白質(zhì)磷酸化的相關(guān)研究起步于20世紀(jì)初,直到近十年,每年平均仍有1.5萬篇相關(guān)研究被報(bào)道。據(jù)估計(jì),真核生物中有將近1/3的蛋白能夠被磷酸化。人類蛋白質(zhì)組中含有10多萬個(gè)潛在的磷酸化位點(diǎn)。相對(duì)于磷酸化修飾,蛋白的焦磷酸化修飾作為一種新發(fā)現(xiàn)的PTM,研究歷史相對(duì)較短。焦磷酸化修飾最早被發(fā)現(xiàn)于糖原合成中。由于目前沒有蛋白質(zhì)的焦磷酸化酶的相關(guān)報(bào)道,一般認(rèn)為蛋白焦磷酸化屬于無酶催化的修飾。崔雋課題組發(fā)現(xiàn)焦磷酸化酶UAP1可以結(jié)合到轉(zhuǎn)錄因子IRF3上,直接正向調(diào)控I型干擾素反應(yīng),還發(fā)現(xiàn)UAP1作為第一個(gè)被發(fā)現(xiàn)的蛋白焦磷酸化酶,通過催化IRF3的焦磷酸化,增強(qiáng)機(jī)體固有免疫應(yīng)答,降低病毒感染的新機(jī)制。
該研究首先通過功能篩選,發(fā)現(xiàn)敲除糖基化底物合成中關(guān)鍵的焦磷酸化酶UAP1能夠明顯抑制細(xì)胞的抗病毒活性,而過表達(dá)UAP1則會(huì)增強(qiáng)I型干擾素以及干擾素刺激基因的表達(dá)。Uap1敲低的小鼠對(duì)各種病毒的感染都更敏感,其抗病毒能力明顯下降,揭示了其在機(jī)體抗病毒免疫中的重要作用。為了探究UAP1調(diào)控抗病毒免疫的具體機(jī)制,研究人員通過免疫共沉淀實(shí)驗(yàn)發(fā)現(xiàn)UAP1可以特異性結(jié)合轉(zhuǎn)錄因子IRF3。UAP1的酶活突變體喪失了其促進(jìn)I型干擾素通路激活的能力,表明UAP1的抗病毒功能依賴于其酶活性。研究人員進(jìn)一步確定UAP1主要通過催化IRF3的焦磷酸化修飾,而不是糖基化修飾影響IRF3的活性。通過抑制劑實(shí)驗(yàn),研究人員發(fā)現(xiàn)O-糖基化抑制劑(OSMI-1)或者N-糖基化抑制劑(TM)處理細(xì)胞不會(huì)影響UAP1對(duì)I型干擾素的促進(jìn)作用,而焦磷酸化修飾抑制劑(TNP)處理會(huì)明顯抑制UAP1的抗病毒功能。補(bǔ)回UAP1在己糖胺通路中的重要產(chǎn)物UDP-N-乙酰基葡萄糖胺,不能回復(fù)敲除UAP1對(duì)IRF3及其抗病毒免疫的調(diào)控作用,揭示了UAP1可能直接通過催化蛋白的焦磷酸化發(fā)揮功能。進(jìn)一步的研究通過同位素示蹤法、焦磷酸試劑盒檢測(cè)以及質(zhì)譜實(shí)驗(yàn)等不同實(shí)驗(yàn)方法,證明UAP1能夠特異性催化IRF3的Ser386位點(diǎn)的焦磷酸化修飾。研究人員構(gòu)建了Ser386位點(diǎn)失活的S386A突變體,發(fā)現(xiàn)S386點(diǎn)突變后IRF3不能進(jìn)一步活化,IRF3 C端5ST活性位點(diǎn)(Ser396)也不能發(fā)生磷酸化,同時(shí)IRF3二聚化和入核能力也明顯下降,這些結(jié)果表明UAP1介導(dǎo)的焦磷酸化修飾對(duì)抗病毒固有免疫的調(diào)控至關(guān)重要。
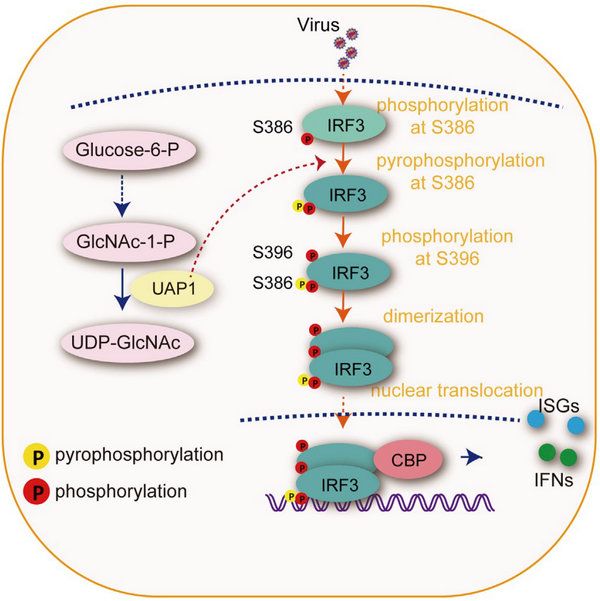
圖2 UAP1通過催化IRF3焦磷酸化,調(diào)控IRF3活性及其I型干擾素信號(hào)通路的工作模型
中山大學(xué)生命科學(xué)學(xué)院崔雋教授為該研究論文的通訊作者,中山大學(xué)生命科學(xué)學(xué)院楊帥博士為第一作者。該研究得到了國(guó)家自然科學(xué)基金等經(jīng)費(fèi)支持。


